12 Regulation, Privacy, and the IRB
The voluntary consent of the human subject is absolutely essential.
The Nuremberg Code (1947)
12.1 Learning objectives
By the end of this chapter you should be able to:
- Reason about FDA regulation of AI Software as a Medical Device (SaMD), including predetermined change control plans (PCCPs) for adaptive AI.
- Identify when an AI use case falls under EU AI Act high-risk medical classification and what compliance obligations follow.
- Draft IRB protocol language that addresses AI-assisted analysis, including data flow, vendor agreements, and audit logging.
- Apply HIPAA in the LLM era: business associate agreements (BAAs), zero-data-retention configurations, and on-prem versus API tradeoffs for protected health information.
12.2 Orientation
The regulatory landscape for AI in health is moving faster than any textbook chapter can keep up with. The specifics in this chapter are accurate as of early 2026 and will date. The conceptual structure, what kinds of AI uses fall under what regulatory regimes, what documentation is expected, is more stable and is the emphasis of the chapter.
The researcher’s role in the regulatory side of AI work is often underestimated. The role is not to be the regulatory expert (that is the regulatory affairs team’s job) but to ensure the analysis is conducted in a way that supports the regulatory work. A researcher who designs an AI-assisted study without considering the SaMD classification, the IRB language, or the HIPAA flow may produce defensible analytical work that cannot be used without rework.
The chapter develops four threads. FDA SaMD and PCCPs: when an AI tool is a regulated medical device and what predetermined change control plans add. EU AI Act: the high-risk medical classification and what compliance obligations follow for products with EU exposure. HIPAA in the LLM era: business associate agreements with API providers, zero-data-retention configurations, on-prem alternatives. IRB language for AI-assisted protocols: what to write so the review board has what it needs.
12.3 The frameworks at a glance
Before we work through the individual regimes, it helps to see them side by side, because a common source of confusion is treating them as interchangeable when they differ in what they govern, in whether they carry the force of law, and in where they apply. Table 12.1 lays out the five frameworks a health researcher is most likely to encounter. The reader may treat the final column as a working rule: for most population-health research the binding regimes (FDA SaMD, EU AI Act) do not apply, and the useful material is in the voluntary frameworks that supply documentation structure.
| Framework | What it governs | Binding vs guidance | Geographic scope | Relevance to a public-health researcher |
|---|---|---|---|---|
| FDA SaMD / PCCP (U.S. Food and Drug Administration, 2024) | Software that is a medical device (diagnosis, treatment, triage) | Binding (federal law) | US market | Applies only when a tool informs clinical action; most population research is out of scope, but a surveillance signal that triggers care can edge in |
| EU AI Act (European Parliament and Council, 2024) | AI systems by risk tier; most medical AI is ‘high-risk’ | Binding (EU regulation) | EU market or use | Triggers with EU deployment or EU subjects; its data-governance and bias-audit duties bear on equity work |
| WHO LMM guidance (World Health Organization, 2024) | Ethics and governance of large multimodal models in health | Guidance (non-binding) | Global / advisory | Frames population-level and lower-resource-setting equity concerns; useful for global-health and surveillance framing |
| NIST AI RMF (National Institute of Standards and Technology, 2023) | A risk-management process (govern, map, measure, manage) | Voluntary framework | US-origin, used internationally | A practical scaffold for documenting risk in a protocol regardless of sector |
| CHAI | Voluntary assurance standards and reporting (model cards, assurance labs) | Voluntary consensus | US-based consortium | Emerging institutional reporting norms you may be asked to follow |
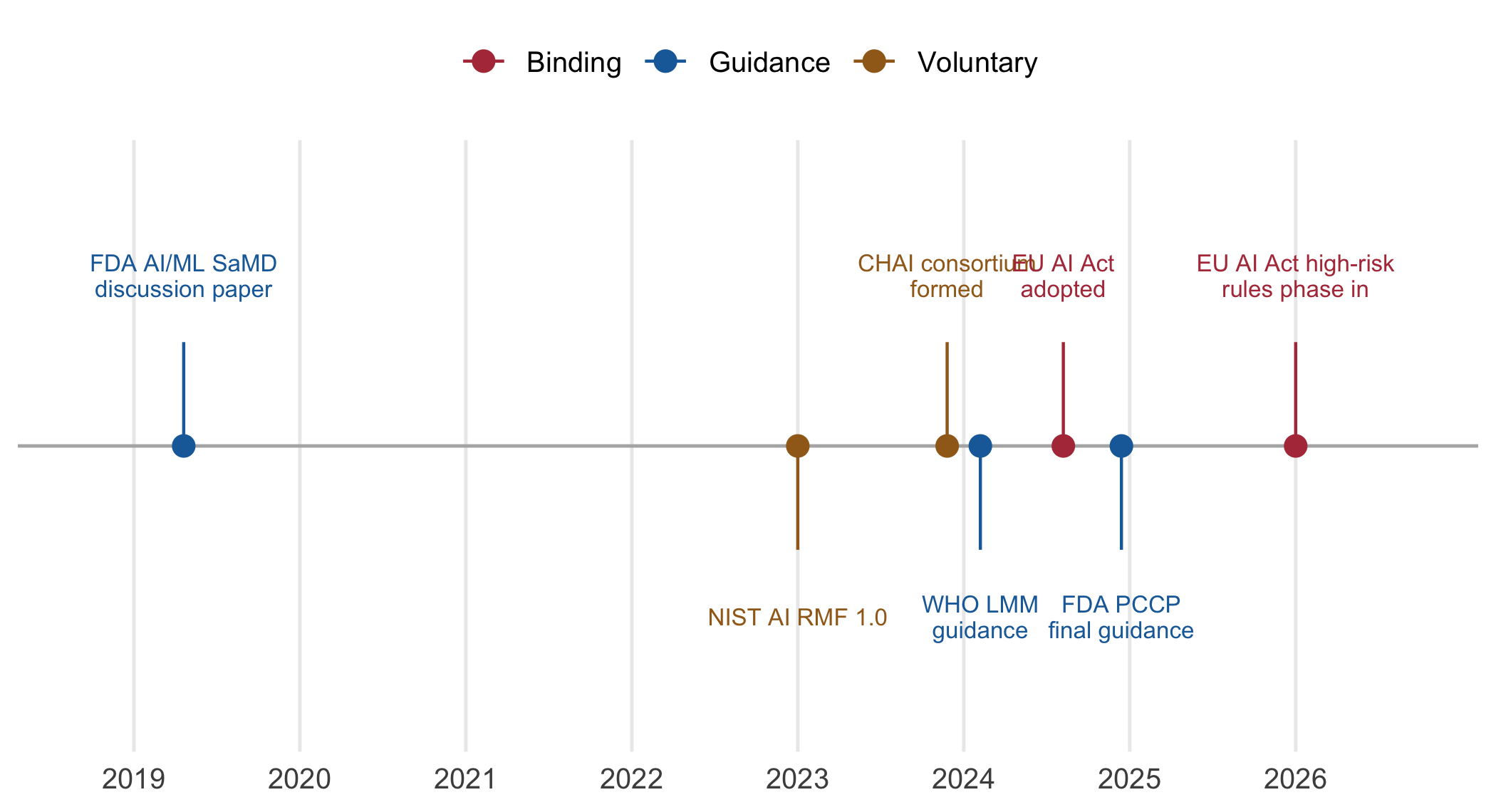
These frameworks did not arrive together. Figure 12.1 traces the milestones since 2019, and the reader should take from it that the binding instruments (in red) are recent and still phasing in, whereas the guidance and voluntary frameworks (blue and gold) arrived first and remain the practical reference for research that is not a regulated product.
12.4 The researcher’s contribution
Three judgments are not delegable.
(Judgment 1.) Identifying when a tool is a medical device. A model that produces a clinical decision: ‘this patient is high risk for sepsis’, that informs clinician action is potentially regulated by the FDA as SaMD. A model used for research-only analysis is typically not. The line is consequential: SaMD classification triggers substantial regulatory burden; research-only does not. The researcher identifies where on the line a project sits and ensures the appropriate framing is documented.
(Judgment 2.) Knowing the data-flow path. Patient data may flow through multiple parties in an AI- assisted analysis: the institution, an API provider, possibly a hosted RAG corpus, possibly a logging service. Each hop needs to be HIPAA-compliant or the analysis cannot proceed with PHI. The researcher maps the data flow, confirms the BAAs are in place, and documents the configuration.
(Judgment 3.) Drafting IRB language that survives review. IRB protocols for AI-assisted research need to address questions the IRB will ask: what AI is being used, what data flows where, what verification is performed, what subject-level risks attach to the AI use. The researcher drafts this language so the IRB’s review is informed and the protocol is approvable.
These judgments are what distinguish AI-assisted work that proceeds from work that is held up at IRB or regulatory review.
The three judgments can be arranged as a short decision path, which is worth internalising because it determines which regime attaches before any project work begins. Figure 12.2 walks the reader from the first question, is this research or is it clinical care, through to the frameworks that apply at each terminal. We note that most population-health research lands on the left branch, where the IRB and HIPAA govern and the binding device regimes do not.
flowchart TD A["Is the activity research,<br/>or clinical care / product deployment?"] A -->|Research| B["Does it involve<br/>human-subjects data?"] A -->|Clinical care<br/>or product| C["Does the tool inform<br/>a clinical decision?"] B -->|Yes| D["IRB review<br/>+ HIPAA data-flow map"] B -->|"No, or public / aggregate data"| E["Likely IRB-exempt;<br/>still map the data flow"] C -->|"Yes: diagnosis,<br/>treatment, triage"| F["Likely FDA SaMD<br/>+ EU AI Act high-risk"] C -->|"No: admin,<br/>retrieval, education"| G["Outside SaMD;<br/>governance guidance applies"] D --> H["Frameworks in play:<br/>HIPAA, NIST AI RMF,<br/>WHO LMM, CHAI"] E --> H G --> H F --> I["Pre-market pathway<br/>+ PCCP for adaptive AI"]
12.5 FDA SaMD and predetermined change control plans
The FDA regulates Software as a Medical Device when the software’s intended use places it within the device definition. For AI, this typically means:
Diagnostic AI: the model produces a diagnostic output that is presented to a clinician.
Treatment recommending AI: the model recommends a specific treatment.
Workflow-prioritizing AI in safety-critical contexts: e.g., a model that prioritizes ED triage, stroke detection on imaging, sepsis alerts.
Tools that are clearly outside SaMD include: research-only AI not used in clinical care, administrative AI (scheduling, documentation assistance), medical-education AI, and pure information-retrieval AI without recommendation output.
The classification matters because SaMD imposes substantial regulatory requirements: pre-market clearance or approval (510(k), De Novo, PMA depending on risk class), quality management system, post-market surveillance, adverse-event reporting. Pre-market timeline is months to years.
Predetermined Change Control Plans (PCCPs) (U.S. Food and Drug Administration, 2024) are an FDA mechanism specifically for AI/ML SaMD. Under a PCCP, the manufacturer specifies in advance what changes the model can undergo without new FDA review (e.g., periodic re-training on updated data, certain types of fine-tuning) and the verification protocols that accompany each. The PCCP trades flexibility (faster updates) for upfront work (specifying the change envelope and the verification in advance).
For a researcher supporting a SaMD development:
- Understand whether the project is targeting SaMD classification or is research-only.
- For SaMD: contribute the analytical validation data the FDA submission requires (performance metrics, subgroup analyses, comparison to standard of care).
- For SaMD with PCCPs: contribute to specifying the change envelope (what re-training is in scope) and the verification regime (what tests confirm the re-trained model is acceptable).
- For research-only: ensure the documentation makes the research-only intent unambiguous.
The published guidance on AI/ML in SaMD has been evolving since 2019 and the specifics will continue to change. The conceptual framework, risk-based regulation, life-cycle approach, transparency expectations, is stable.
12.6 EU AI Act and high-risk medical classification
The EU AI Act (European Parliament and Council, 2024) applies to any AI system placed on the market or used in the EU. It classifies AI systems by risk level: prohibited, high-risk, limited- risk, minimal-risk. Most medical AI falls into high-risk, which triggers substantial compliance obligations.
High-risk AI in the medical domain includes:
- AI as part of a regulated medical device (most clinical AI).
- AI used for triage of patients in healthcare facilities.
- AI used for hiring or promotion decisions in healthcare staffing (separately).
High-risk AI obligations include:
- Risk management system.
- Data governance (training-data documentation, bias mitigation).
- Technical documentation.
- Record-keeping (logging of operation).
- Transparency to users.
- Human oversight.
- Accuracy, robustness, cybersecurity.
- Conformity assessment.
For a researcher on a clinical-AI project with EU exposure (deployment in EU or research collaboration with EU institutions), these translate into:
- Document the data sources and the data-quality measures used.
- Audit for subgroup fairness (the EU AI Act has explicit non-discrimination expectations).
- Specify the human-oversight model (who reviews, what they review, how they override).
- Maintain an audit log of model operations.
The AI Act is being phased in through 2026; the specifics of high-risk-system compliance are still being clarified through implementing regulations. As with FDA SaMD, the conceptual structure is stable.
12.7 HIPAA in the LLM era
The Health Insurance Portability and Accountability Act governs the use and disclosure of protected health information (PHI) in the US. The LLM era introduces specific concerns.
API providers as business associates. When PHI is sent to an LLM API (Anthropic, OpenAI, Google), the API provider is a business associate of the covered entity. A Business Associate Agreement (BAA) must be in place. The major API providers offer HIPAA-eligible configurations with BAAs, but the configurations are specific (not all endpoints, not all features) and require contractual setup.
Zero-data-retention configurations. API providers support ‘zero retention’ modes where requests and responses are not persisted on the provider’s infrastructure. For PHI use, this is the appropriate configuration. Verify the configuration is in place before sending PHI; the default is often retention.
On-prem alternatives. For organizations that cannot or will not use API providers, on-prem deployment of open-weights models (Llama family, Mistral, etc.) is an alternative. The trade is substantial: on-prem deployment requires GPU infrastructure, MLOps, and the operational work of maintaining a model. The cost calculus depends on the organization; for high-volume use with sensitive data, on-prem may pencil out; for moderate-volume research use with appropriate BAAs, API is usually operationally simpler.
De-identification before transmission. When the analysis can be performed on de-identified data (HIPAA Safe Harbor or expert-determination), de-identifying before transmission avoids the BAA question entirely. For research that does not require patient-level identifiers, this is the cleanest path.
A practical decision tree:
Does the analysis require PHI?
├── No → De-identify, transmit freely
└── Yes
├── Can you use an API with BAA + zero-retention?
│ └── Yes → Configure, document, proceed
└── No → On-prem deployment
└── Justify the cost, deploy, maintainFor most applied research, de-identification or BAA+API is sufficient. On-prem is reserved for specific contexts (high-volume clinical operations, specific contractual constraints).
12.8 Population data, de-identification, and the surveillance exemption
Much of public-health research works with population data rather than individual clinical decisions, and the governance questions shift accordingly. We note three points that are specific to this setting and are easy to get wrong.
First, de-identification is harder for free text than the Safe Harbor list suggests. HIPAA Safe Harbor enumerates eighteen identifiers to remove, and for structured registry fields their removal is mechanical. Clinical free text is another matter: a chief-complaint note or a discharge summary carries quasi-identifiers (an unusual occupation, a rare diagnosis, a named facility) that Safe Harbor does not enumerate and that a capable language model can surface or recombine. Unfortunately, running such text through an LLM does not de-identify it; if anything the model’s fluency with context raises the re-identification risk. Where the text itself must be processed, expert determination, rather than Safe Harbor, is generally the defensible route.
Second, the public-health surveillance exemption governs collection and use, not the vendor hop. The HIPAA Privacy Rule permits a covered entity to disclose PHI to a public health authority for surveillance without individual authorization (the public-health-activities provision), and the Common Rule treats public-health surveillance as an activity that is not research, so the IRB requirement may not attach at all. But how far does that exemption reach? It authorizes the surveillance itself; it says nothing about sending the data through a third-party API. That hop still needs a BAA, a zero-retention configuration, or prior de-identification. In sum, the exemption removes the IRB question, not the data-flow question.
Third, IRB and ethics review for LLM studies on population data attends to group-level harm, not only individual re-identification. When a model classifies or summarizes data on small subgroups (a rare condition in a specific county, an immigrant community, an occupational cohort), the output can enable stigmatising or discriminatory inference about the group even when no individual is identified. The WHO guidance on large multimodal models (World Health Organization, 2024) makes this population-level and equity framing explicit, and it is the framing a public-health IRB will expect.
To illustrate, consider a syndromic-surveillance team that wants to use an LLM to code free-text emergency-department chief complaints into syndrome categories (influenza-like illness, gastrointestinal, heat-related) for a weekly outbreak-monitoring dashboard. The surveillance activity is covered by the public-health exemption, so the IRB question is light. The frameworks that do the work here are the ones on the left branch of Figure 12.2: HIPAA governs whether the free text can leave the perimeter, and the governance frameworks structure the documentation. The team de-identifies by expert determination before transmission, uses a BAA-covered zero-retention endpoint, validates the coding against a manual sample, and, following the NIST AI RMF’s measure-and-manage steps (National Institute of Standards and Technology, 2023), records the classifier’s error rate by syndrome and by facility so that a systematic miss in one community is caught rather than propagated into the dashboard.
12.9 IRB language for AI-assisted protocols
IRBs increasingly review protocols that include AI- assisted analysis. The questions they ask are predictable; the language that addresses them is relatively standard.
Disclosure of AI involvement. What AI is being used and for what purpose. ‘A reasoning model from [provider] is used to assist with literature review, analysis planning, and methods drafting’ is the standard language.
Data flow. What data goes where. A diagram or narrative covering: data extraction (from EHR or registry), de-identification (Safe Harbor or expert determination), transmission (which API or on-prem), return path (output back to investigator), storage (local file system, cloud, etc.).
Subject-level risk. Whether AI involvement creates risks for subjects beyond standard data handling. For de-identified data with appropriate API configurations, the answer is usually ‘no additional risk beyond standard data handling’. For PHI use, the answer addresses the BAA, zero-retention, and audit configuration.
Verification regime. What human review or verification accompanies the AI assistance. ‘All AI-generated text in publications is reviewed by the investigator before submission. AI-generated code is verified by independent execution against test data.’
Audit logging. Whether and how AI operations are logged. For research with regulatory implications, an audit log of every API call (timestamp, prompt, response, version) is increasingly expected.
A working pattern: a 1-page ‘AI Assistance and Verification Plan’ as an appendix to the protocol. The IRB review proceeds with this in hand; questions are answered up-front rather than through reviewer back-and-forth.
12.10 Worked example: an AI-assisted observational study
A research team wants to use AI assistance for a retrospective observational study using the institutional EHR. The study examines outcomes after a specific procedure across the past 5 years (~3,000 patients). AI assistance will be used for:
- Literature review (deep research).
- Phenotype identification from clinical notes (LLM-based extraction).
- Statistical analysis planning (reasoning model).
- Methods drafting.
Step 1: classify the AI uses. None of the uses produces clinical recommendations or diagnostic outputs. The study is research-only; SaMD does not apply. EU AI Act does not apply (no EU subjects, no EU deployment). The relevant frameworks are HIPAA and the IRB.
Step 2: data flow. EHR extraction is de-identified by the team’s honest broker before release. The de-identified dataset is transmitted to an API provider (Anthropic, with BAA + zero- retention configured). Responses are stored locally; no PHI ever leaves the institutional perimeter.
Step 3: HIPAA confirmation. The IT and compliance teams confirm the BAA is in place, the API configuration is zero-retention, and the workflow log captures all transmissions. Documentation is filed with the data-use agreement.
Step 4: IRB protocol language. A 1-page appendix to the protocol covers:
AI Assistance and Verification Plan
This study uses generative AI assistance for the
following purposes:
1. Literature review: An AI-assisted scoping review
tool (OpenAI Deep Research) is used for initial
literature search. All cited papers are
manually verified by the investigator before
inclusion.
2. Phenotype extraction from de-identified clinical
notes: A multimodal LLM (Anthropic Claude) is
used to extract phenotype indicators from the
research dataset. Extraction is validated against
manual chart review on a 10% random sample;
discrepancies > 5% trigger re-validation and,
if necessary, re-prompting.
3. Statistical analysis planning: A reasoning model
(Anthropic Claude with extended thinking) assists
in drafting analysis plans. All plans are
reviewed and finalised by the senior
researcher.
4. Methods drafting: AI-generated text is reviewed
and edited by the investigator before publication.
AI involvement is disclosed in the methods
section.
Data flow: De-identified data only is transmitted to
the AI provider. The provider has a Business Associate
Agreement with the institution and is configured for
zero data retention. Audit logs of all transmissions
are maintained for the duration of the study + 6
years.
Subject-level risk: The AI involvement does not
introduce subject-level risks beyond those of the
underlying data handling, which is governed by the
institutional data-use agreement.Step 5: review. The IRB reviews the protocol, asks two clarifying questions about the phenotype-extraction validation, and approves with minor modifications. Total IRB review time: 4 weeks.
Step 6: ongoing. As the study proceeds, the extraction-validation results are tracked. Two discrepancies above the 5% threshold trigger re- validation; in both cases the prompt is refined and extraction is rerun. The audit log captures every API call.
The pattern is reproducible across studies. The appendix is the team’s template; the data-flow diagram is updated per study; the IRB review is informed and timely.
12.11 Collaborating with an LLM on regulation and the IRB
Three prompt patterns illustrate working with LLMs on regulatory work.
Prompt 1: ‘Help me classify this AI use case for regulatory purposes.’ Provide the use case description.
What to watch for. The LLM produces a competent preliminary classification. It tends to oversimplify edge cases. The classification needs sign-off from the regulatory affairs team for any project where classification has material implications.
Verification. Use the LLM’s classification as a starting point for the conversation with regulatory affairs. Do not treat it as definitive.
Prompt 2: ‘Draft IRB language for this protocol’s AI use.’ Provide the protocol summary.
What to watch for. The LLM produces standard language. Verify the specifics, provider names, BAA configurations, validation approaches, match the actual project.
Verification. The IRB language is reviewed by the team’s senior investigators and (where applicable) the institution’s IRB pre-review office.
Prompt 3: ‘Audit this protocol for missing AI-related disclosures.’ Provide the protocol.
What to watch for. The LLM is reasonably good at identifying gaps relative to standard expectations. It may not catch institution-specific requirements; those need to be checked against the IRB’s specific guidance.
Verification. The audit recommendations are reviewed against the IRB’s published guidance and recent decisions.
The meta-pattern: regulatory work is human-authored even when AI-assisted. The LLM accelerates drafting; the responsibility for accuracy and completeness remains with the regulatory affairs team and the investigator.
12.12 Principle in use
Three habits define defensible work in this area:
Classify the use case before designing the project. The classification (SaMD or research, high-risk-AI-Act or not, PHI or de-identified) determines what the project must include from the start. After-the-fact classification produces re-work.
Document the data flow before transmission. Map where PHI goes, confirm BAAs are in place, configure zero-retention. Documentation is easier before the first transmission than after.
Pre-draft the IRB AI-assistance appendix. Maintain the team’s template appendix as a reusable artifact. Each study customises but does not write from scratch.
12.13 Exercises
For an AI-assisted project of yours, write a 1-paragraph SaMD classification: in scope, out of scope, or borderline, with rationale.
Diagram the data flow for a project that uses PHI with an LLM API. Identify each hop and the protection at each.
Draft an IRB AI-assistance appendix for a protocol you are working on. Have a colleague review it.
Read the FDA’s most recent SaMD-AI guidance and summarize it in 2 paragraphs. Compare against what you would have expected.
For a clinical AI deployment in your institution, identify what would change about the deployment if the institution acquired EU exposure.
12.14 Further reading
- U.S. Food and Drug Administration (2024), FDA Guidance on Predetermined Change Control Plans for AI/ML SaMD. The reference for adaptive AI as a medical device.
- European Parliament and Council (2024), EU Artificial Intelligence Act. The regulatory text and accompanying guidance.
- World Health Organization (2024), WHO Guidance on Ethics and Governance of Large Multi-Modal Models in Health. The reference for population-level and equity framing.
- National Institute of Standards and Technology (2023), NIST AI Risk Management Framework. A voluntary, sector-neutral scaffold for documenting risk.
- Gebru et al. (2021), Datasheets for Datasets. Adjacent: data documentation as a regulatory expectation.
- The HHS Office for Civil Rights HIPAA guidance is the authoritative source on HIPAA in healthcare; read the most recent guidance specifically addressing AI and cloud computing.